Terug naar het richtlijnenoverzicht
Genetische nieraandoeningen en aangeboren urinewegafwijkingen zijn frequente oorzaken van nierfalen in de Nederlandse populatie, met een aandeel van in totaal 26% (Tabel 1). Met name in de groep patiënten die bij aanvang van de nieraandoening jonger is dan 40 jaar, zonder etiologische of classificerende diagnose, is het aannemelijk dat erfelijke nieraandoeningen en urinewegafwijkingen nog een aanvullend percentage verklaren.
Tabel 1: Primaire nieraandoening, diagnoses in RENINE registratie (dd. januari 2015)
Op basis van oude ERA-EDTA diagnosecodering, uitgezet tegen leeftijd ten tijde van eerste nierfunctievervangende therapie, bij patiënten geboren na 1950. (Zie voor de individuele aandoeningscodes die gebruikt zijn voor de indeling van de categorieën bijlage 6).
|
Leeftijd (in jaren) van eerste nierfunctie vervangende therapie |
0t/m9 |
10t/m19 |
20t/m29 |
30t/m39 |
40t/m49 |
50t/m59 |
>60 |
Eindtotaal |
|
Aangeboren/ erfelijk |
29% |
21% |
10% |
10% |
17% |
16% |
11% |
15% |
|
Waarschijnlijk aangeboren urinewegafwijking |
33% |
30% |
17% |
10% |
5% |
2% |
2% |
11% |
|
Onbekend |
5% |
14% |
20% |
17% |
14% |
13% |
8% |
15% |
|
Door hypertensie (onwaarschijnlijk bij jonge patiënten) |
0% |
1% |
5% |
10% |
12% |
12% |
11% |
9% |
|
Andere oorzaken |
33% |
34% |
48% |
54% |
52% |
57% |
67% |
51% |
|
Eindtotaal |
100% (n=452) |
100% (n=977) |
100% (n=2062) |
100% (n=2593) |
100% (n=3067) |
100% (n=2312) |
100% (n=242) |
100% (n=11705) |
Het stellen van een genetische diagnose kan vele belangrijke implicaties hebben. Afdelingen Klinische Genetica zijn goed geoutilleerd om de consequenties voor de patiënt en familieleden te bespreken. Ten eerste kan een genetische diagnose consequenties hebben voor de individuele patiënt: bijvoorbeeld wat betreft behandelopties, inschatten van prognose, indicatie voor extrarenale follow-up, eventueel uitbreiden van transplantatiescreening voor familietransplantatie, bespreken van reproductieve keuzes bij kinderwens. Ten tweede kan een genetische diagnose implicaties hebben voor familieleden: zoals presymptomatische (DNA) diagnostiek en indien mogelijk preventieve behandeling bij bijvoorbeeld Alport syndroom. Ten slotte kunnen er implicaties zijn voor de medische wetenschap en zorg: denk aan voortgang der wetenschap, verbeterde diagnostische classificatie, etc.
Het is belangrijk om te weten dat de klinisch geneticus niet altijd DNA-diagnostiek inzet. Een patiënt kan ook verwezen worden om alleen de voor- en nadelen van genetisch onderzoek te bespreken,
In het geval van kinderwens helpt een genetische diagnose bij erfelijkheidsadvisering, wat betreft het overervingspatroon en/of het inschatten van herhalingskans. Diagnose en herhalingskans tezamen geven richting aan de noodzaak tot advies van uitgebreid echo-onderzoek in een zwangerschap en postnatale follow-up, van mogelijkheden voor prenatale diagnostiek en preïmplantatie genetische diagnostiek (embryoselectie; beide alleen mogelijk met bekende pathogene mutatie(s) in de probandus/indexpatiënt), en van de alternatieve mogelijkheden voor het inrichten van kinderwens, met name sperma-/eiceldonatie, adoptie, afzien van kinderwens.
Het belang van kennis rondom de primaire diagnose geldt niet alleen voor nieuwe patiënten, maar zeker ook voor patiënten uit nefrologische controlepopulaties (zonder duidelijke diagnose) en patiënten die reeds eindstadium nierfalen bereikt hebben.
Deze handreiking is bedoeld voor nefrologen en internisten die betrokken zijn bij de behandeling van patiënten met nieraandoeningen. Met de toenemende mogelijkheden en kennis verandert de plaats van genetisch onderzoek in de klinische praktijk snel. Hierdoor zijn er nauwelijks ‘evidence based’ aanbevelingen te geven ten aanzien van de opsporing van genetische nieraandoeningen. Deze handreiking poogt een toepasbaar overzicht te geven van de mogelijkheden van nefrogenetische diagnostiek en doet suggesties ten aanzien van detectie, vervolgdiagnostiek en verwijsindicaties bij patiënten met een primaire nieraandoening met mogelijk een genetische component.
Namens de NFN richtlijnencommissie verantwoordelijk:
Samenstelling werkgroep
Fotografie
Kritisch gelezen en van suggesties voorzien door:
In de literatuur worden indelingen aangehouden om structuur te brengen in de veelheid van nieraandoeningen met genetische etiologie. Zo kunnen ziektebeelden ingedeeld worden naar de overervingsvorm. Ook kan gekozen worden voor een patiëntgeoriënteerde benadering gebaseerd op de manier waarop de patiënt zich klinisch presenteert, of een meer pathofysiologische benadering gebaseerd op de anatomische locatie en/of histopathologische afwijkingen.
Het is van belang om onderscheid te maken tussen congenitale nieraandoeningen en nieraandoeningen met een genetische oorzaak. Van congenitale nieraandoeningen is sprake als de functionele of morfologische afwijkingen van de nier al voor, bij of kort na de geboorte aanwezig zijn (hoewel het kan zijn dat deze zich pas op latere leeftijd openbaren). Nieraandoeningen met een genetische achtergrond kunnen bij de geboorte reeds herkenbaar zijn, maar kunnen ook op latere leeftijd gediagnosticeerd worden.
Een eerste indeling kan gemaakt worden op basis van de overerving; de aanwezigheid van een bepaald overervingspatroon kan helpen bij de identificatie van een genetische nieraandoening. Er zijn grote conceptuele verschillen tussen monogeen-genetische aandoeningen en polygeen-genetische of multifactoriële aandoeningen (zie bijlage 1).
Er hoeft niet per definitie sprake te zijn van Mendeliaanse (=monogene) overerving om wel diagnostiek naar monogene aandoeningen te verrichten. Een genetische aandoening betekent namelijk dat er vaak -maar niet altijd!- meer mensen in de familie zijn aangedaan. Het is uiteraard zinvol om een familieanamnese af te nemen (zie paragraaf 4).
Er zijn verschillende overervingsvormen (zie NVN Brochure Erfelijkheid voor voorbeelden van verschillende overervingswijzen in een stamboom), namelijk:
In bepaalde gevallen kan incomplete penetrantie of non-penetrantie een rol spelen. Dit betekent dat sommige patiënten die de mutatie dragen toch geen klinische verschijnselen (=fenotype) laten zien. Bij variabele expressielaten mutatiedragers verschillende fenotypes in aard en ernst zien. Hierdoor kan het overervingspatroon soms lastig te herkennen zijn, zoals bijvoorbeeld bij HNF1B mutatiedragers.
Naast de indeling naar overervingspatroon kan de (oorspronkelijke) klinische presentatie van de patiënt verder helpen bij het maken van de genetische differentiaaldiagnose. Belangrijke hoofdgroepen hierbij zijn (2):
Zeker bij patiënten met verder gevorderde nierinsufficiëntie kan het achterhalen van het oorspronkelijke presentatiepatroon lastig zijn. In bijlage 2 zijn tabellen te vinden waarin genetische nieraandoeningen zijn ingedeeld in groepen met deze klinische presentaties.
Een indeling op basis van histopathologisch niveau kan ook gebruikt worden om de differentiaaldiagnose (verder) te verkleinen.(3) Hierbij valt te denken aan verschillende erfelijke vormen van FSGS (4) of tubulo-interstitiële fibrose (5).
Het is belangrijk te weten dat met de toename van kennis duidelijk wordt dat van oudsher bekende nieraandoeninggenen meerdere nefrologische beelden kunnen veroorzaken. Voorbeelden hiervan zijn COL4A3/4/5 (klassiek bij Alport syndroom) en PAX2 mutaties (klassiek bij renaal coloboom syndroom) bij FSGS, en DGKEmutaties bij aHUS. (5,6)
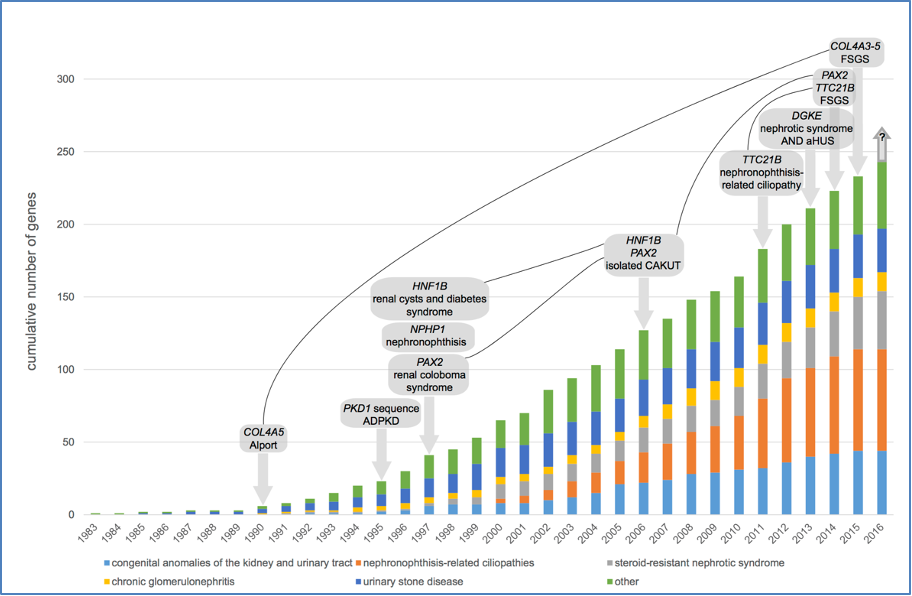
Figuur 1 – Historisch overzicht van aantal geïdentificeerde genen dat een erfelijke nieraandoening veroorzaakt, met nefrogenetische ‘mijlpalen’. Inclusief voorbeelden van uitbreiding van het fenotypisch spectrum van een aantal klassieke nieraandoeninggenen (gennaam cursief, bijbehorend aandoeningbeeld eronder). Het staafdiagram geeft het cumulatieve aantal bij nieraandoening betrokken genen over de jaren, gebaseerd op Vivante en Hildebrandt (6) en de diagnostische ervaring van de auteurs. Publicatiejaartallen zijn gebaseerd op OMIM (https://www.ncbi.nlm.nih.gov/omim).
ADPKD, autosomal dominant polycystic kidney disease; aHUS, atypical hemolytic uremic syndrome; CAKUT, congenital anomalies of the kidney and urinary tract; FSGS, focal segmental glomerulosclerosis.
Figuur overgenomen uit Van Eerde et al 2016 (7) met toestemming van Kidney International.
Hierbij dient opgemerkt te worden dat met de snel uitbreidende kennis over de genetische bijdrage aan nieraandoeningen de overzichten met aandoeningen per definitie gedateerd zijn.(8,7) De tabellen in bijlage 2 dienen ter illustratie en zijn zeker niet volledig. De differentiaaldiagnose kan richting geven aan het genetisch onderzoek dat men inzet. Ook als het klinisch niet goed mogelijk is om een duidelijke richting aan de differentiaaldiagnose te geven, kunnen de moderne uitgebreide genetische onderzoeken, in samenspraak met een klinisch geneticus, indien gewenst uitkomst bieden.
In deze praktische handreiking kunnen we, mede door de snelle ontwikkelingen, geen sluitende genetische differentiaaldiagnoses geven, maar wel algemene handreikingen voor de “genetische aspecten” van de patiënt met een nieraandoening en dan met name de patiënt met, of bij wie gedacht moet worden aan, een monogene aandoening.
In het vervolg worden eerst aandachtspunten ten tijde van het consult belicht, daarna volgen verwijsindicaties en enkele bijlagen met relevante informatie. Omdat het veld zeer in beweging is en het reëel is om te verwachten dat in steeds meer patiëntengroepen DNA-onderzoek vooraan in het diagnostisch traject zal worden gestart (genotype-eerst benadering, in plaats van fenotype-eerst benadering), dient dit document regelmatig herzien te worden. In geval van twijfel, aarzel niet om te overleggen met een klinisch geneticus.
Er zijn verschillende momenten waarop er aanleiding kan zijn voor het (opnieuw) verrichten van (familie)anamnese, lichamelijk onderzoek en (genetisch) aanvullend onderzoek.
Voorbeelden hiervan zijn:
Om de kans op een erfelijke ziekte bij een individuele patiënt in te schatten is het afnemen van een familieanamnese door de nefroloog essentieel. Het is te adviseren dit voor elke patiënt in ieder geval éénmaal te doen en daarna regelmatig te herhalen (zie ook paragraaf 3). De informatie uit de familieanamnese biedt aanknopingspunten voor een vermoeden van genetische ziekte, en is tevens ook behulpzaam bij het schrijven van een verwijzing naar een klinisch geneticus.
Een uitgebreide (familie)anamnese betekent tenminste:
Zie ook ‘Aanvullend genetisch onderzoek’ (paragraaf 7).
Nota Bene:
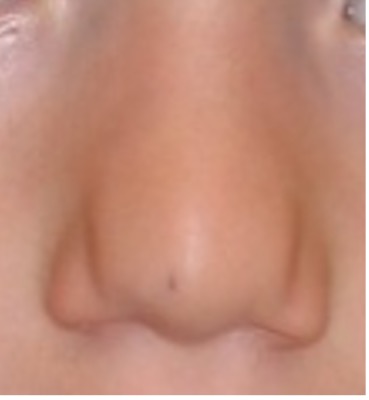
Zoals gebruikelijk dient bij elke patiënt met een nieraandoening een volledig internistisch lichamelijk onderzoek te worden verricht. Hoewel het dysmorfologisch onderzoek bij uitstek de expertise van de klinisch geneticus is, tonen wij hier een aantal dysmorfieën die kunnen voorkomen bij genetische aandoeningen die ook tot nierfunctiestoornissen kunnen leiden en makkelijk herkenbaar zijn (foto’s met dank aan Prof.dr. R.C.M. Hennekam, klinisch geneticus AMC en dr. M.J. v.d. Boogaard, klinisch geneticus UMC Utrecht). Zie voor een overzicht van gestandaardiseerde terminologie: Elements of morphology.
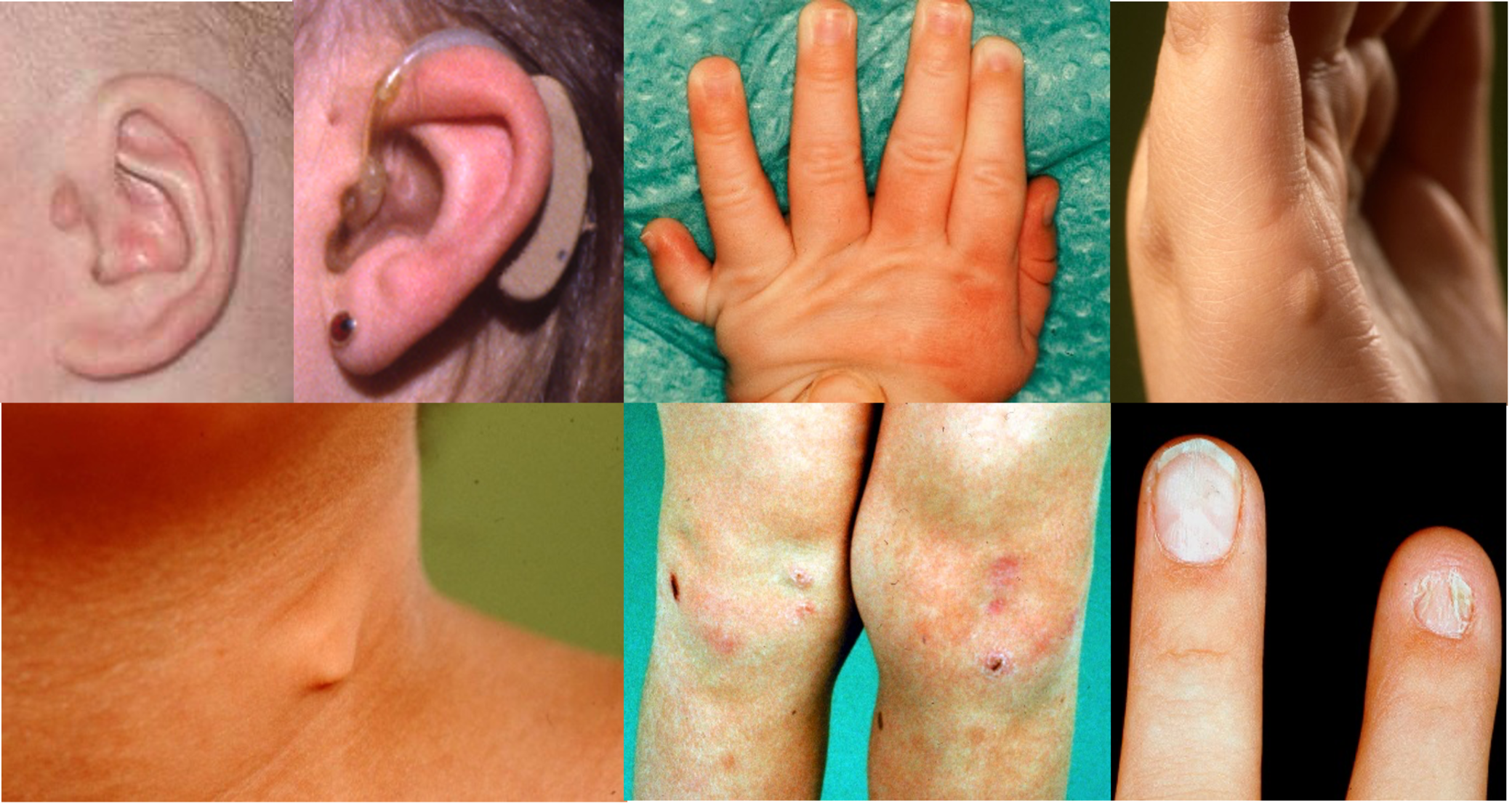
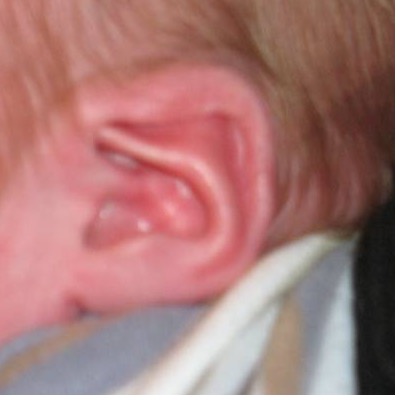
Van links naar rechts bovenste rij: Afwijkende vorm van het oor (o.a. Townes-Brocks syndroom; bij patiënt op de foto ook een preauriculaire tag), preauriculaire pit (brachio-oto-renaalsyndroom (BOR), postaxiale polydactylie, inclusief postminimus of litteken na verwijdering zoals op de foto uiterst rechts (Bardet-Biedl syndroom en overige ciliopathieën).
Van links naar rechts middelste rij: Halsfistel (BOR), kleine of afwezige patellae en afwijkende nagels (beide passend bij nagel-patella syndroom).
Onderste rij: kleine, wat vierkante oren en opvallende neus met kleine alae nasi (22q11.2 deletie syndroom)
Enkele niet getoonde afwijkingen zijn: colobomen (NB bij het renaal coloboomsyndroom geen iris of rentina coloboma maar juist n. opticusafwijkingen), triphalangeale duim (Townes-Brocks syndroom) en enkele huidafwijkingen zoals angiokeratomen (ziekte van Fabry) of fibrofolliculomen (Birt-Hogg-Dubé syndroom).
Aanvullend onderzoek kan helpen het klinisch beeld verder in kaart te brengen. De waarde van de onderzoeken is niet alleen afhankelijk van de differentiaaldiagnose maar ook van de timing van de diagnostiek. Als men bij vergevorderd nierfalen op zoek wil naar de primaire diagnose kan het zinvol zijn om de data rondom de eerste presentatie te achterhalen. Toch kan aanvullend onderzoek ook bij gevorderde nierinsufficiëntie zinvol zijn, denk aan beeldvorming van de nieren op zoek naar aanlegstoornissen of opvallende metabole stoornissen die niet verklaard kunnen worden door gevorderde nierinsufficiëntie.
Naast een gebruikelijk lichamelijk en aanvullend onderzoek, kan breder niet-genetisch aanvullend onderzoek de differentiaaldiagnose meer richting geven. Denk bijvoorbeeld aan urinezuur en magnesium bij nierinsufficientie en cysten (mogelijk HNF1B-gerelateerde ziekte).
Bij DNA-onderzoek kan er gekozen worden uit één gerichte test voor één specifiek gen (Sanger sequencing), maar ook voor een panel genen bestaande uit meerdere genen die alle geassocieerd zijn met een groep aandoeningen met een vergelijkbaar klinisch fenotype of bijvoorbeeld voor alle genen waarvan bekend is dat zij geassocieerd zijn met nieraandoeningen. Uiteraard zijn exoom-/genoombrede (‘whole exome’ of ‘whole genome’) sequencing tests ook mogelijk. Vaak worden genpanels gefilterd van whole exome data, in dat geval is het mogelijk om in een later stadium een volgende analyse van de bestaande data te verrichten. Zie voor verdere uitleg onderstaand kader met terminologie.(11)
Terminologie
Genetische diagnostiek van enkele genen of kleinere fenotypegerichte genpanels kan, maar hoeft niet, door een goedgeïnformeerde nefroloog zelf aangevraagd worden. Iedere (kinder)nefroloog is bevoegd om DNA-onderzoek aan te vragen bij patiënten met symptomen van een erfelijke ziekte, waarbij bekwaamheid met betrekking tot pre- en posttest counseling en vertrouwdheid met de interpretatie van de uitslagen doorslaggevend zijn. Voorafgaand aan het inzetten van genetisch onderzoek is het essentieel de implicaties die genetische diagnostiek kan hebben in overweging te nemen (zie ook paragraaf 7.3 en 7.6) en mee te nemen in de beslissing wel of geen genetisch onderzoek in te zetten. Goedgeïnformeerd houdt in dat de nefroloog al het onderstaande beheerst:
(zie ook paragraaf: Verwijsindicaties)
Een patiënt kan uiteraard altijd verwezen worden vanwege een verdenking op een genetische aandoening, bijvoorbeeld om genetisch aanvullend onderzoek in het kader van genetische counseling plaats te laten vinden (zie ook paragraaf: Verwijs-/overlegredenen: nog niet gestelde genetische diagnose). Tevens kan laagdrempelig overlegd worden met een multidisciplinair expertisecentrum, waar ingeschat kan worden of een verwijzing geïndiceerd is en zo ja of deze het beste naar een gespecialiseerde nefroloog of direct naar een klinisch geneticus of naar een combinatiespreekuur kan zijn. Een verwijzing naar een klinisch geneticus verdient de voorkeur in de volgende gevallen:
De timing van genetische diagnostiek kent twee aspecten: aan de ene kant de plaats van genetische diagnostiek in het uitwerken van de differentiaaldiagnose, en aan de andere kant de patiëntfactoren die aanleiding kunnen geven tot het verrichten van genetisch onderzoek.
Waar genetische diagnostiek voorheen met name werd verricht nadat de klinische diagnose praktisch al was gesteld, zijn er steeds meer redenen om de genetische DNA-diagnostiek naar voren te halen in het diagnostische traject:
De verschillende beweegredenen om DNA-diagnostiek te verrichten zijn ook nauw verbonden met de timing van deze diagnostiek in het leven van patiënten:
DNA-diagnostiek waarvan de uitslagen gebruikt moeten kunnen worden als basis voor prenatale diagnostiek, preïmplantatie genetische diagnostiek of presymptomatische diagnostiek kan enkel worden verricht in gecertificeerde DNA-laboratoria; de meeste zijn verbonden aan de academische centra.
Voor gerichte gendiagnostiek kan nagekeken worden op www.DNAdiagnostiek.nl welke centra een specifieke test verrichten. Via deze site zijn ook de aanvraagformulieren te downloaden. Door de snelle ontwikkelingen lopen de aanvraagformulieren op de website nog wel eens iets achter op het actuele aanbod. Bij twijfel kunt u overleggen. U kunt overleggen met de laboratoriumspecialist klinische genetica verantwoordelijk voor de specifieke test (telefoonnummer via www.DNAdiagnostiek.nl), bij meer klinische vragen kunt u overleggen met een klinisch geneticus.
Zodra de uitslag bekend is wordt deze alleen naar de aanvrager (en eventuele kopiehouders) verstuurd. Andere behandelaren kunnen deze uitslag alleen met expliciete toestemming van patiënt opvragen (telefoonnummers via www.DNAdiagnostiek.nl).
Na het onderzoek blijft het overgebleven DNA tenminste 30 jaar- hoewel in de praktijk de levensduur van 3 generaties (~100 jaar) wordt aangehouden- bewaard in het betreffende laboratorium en kan gebruikt worden voor vervolgdiagnostiek en/of wetenschappelijk onderzoek indien de patiënt daar schriftelijke toestemming voor geeft. Er is een procedure om het materiaal te vernietigen indien gewenst. De aanvraag en de uitslagbrief dienen 115 jaar bewaard te worden.
Counseling door een klinisch geneticus en DNA-onderzoek zijn voor de verzekeraar aparte verrichtingen. DNA-onderzoek valt buiten de DOT-structuur. Beide behoren tot het basispakket, maar gaan wel ten koste van het eigenrisicobedrag (https://www.umcutrecht.nl/nl/Ziekenhuis/Afdelingen/Genetica).
Voor het onderzoeken van één gen wordt een standaardtarief van ongeveer €800,- in rekening gebracht. Voor het verrichten van NGS-diagnostiek wordt het dubbele standaardtarief gerekend. Gericht DNA-onderzoek naar een al in de familie bekende DNA-variant is goedkoper dan een enkel standaardtarief (prijspeil 2017).
Een (presymptomatische) diagnose kan consequenties hebben voor het afsluiten van verschillende verzekeringen. Er wordt in Nederland onderscheid gemaakt tussen te verzekeren bedragen onder en boven de zogenoemde ‘vragengrens’. De vragengrens ligt voor levensverzekeringen op 268.125 euro, voor het eerste jaar arbeidsongeschiktheid op 38.877 euro (d.d. 2017).
Boven en onder de vragengrens
Moet iemand altijd klachten, verschijnselen en aandoeningen melden (zie voor presymptomatische DNA-diagnoses én een levensverzekering onder de vragengrens, hieronder).
Als iemand gezondheidsproblemen heeft van een erfelijke aandoening is die persoon wettelijk verplicht dit aan de verzekeraar te melden. Als de specialist de symptomen ziet als duidelijke kenmerken van de erfelijke aandoening moet iemand deze symptomen melden aan de verzekeraar. Ook als hij of zij verder nog geen klachten heeft. Of iets wel of niet een kenmerk is van een (erfelijke) aandoening is aan de expertise van een medicus. Zie ook: Melden aan verzekeraar
Bij bedragen onder de vragengrens:
Bij bedragen boven de vragengrens:
Boven de vragengrens mag dit wel worden gevraagd, maar verzekeraars gaan daar verschillend mee om. Het is belangrijk voor patiënten om zich goed te laten informeren maar ook voor dokters die hierin adviseren om op de hoogte te zijn. Patiënten kunnen hierover voorgelicht worden bij een klinisch genetisch centrum. Meer informatie: https://erfelijkheid.nl/special/verzekeren, vanatotzekerheid.nl.
Bij autosomaal dominante aandoeningen die leiden tot eindstadium nierfalen is het – in het kader van gevolgen voor eigen verzekerbaarheid en consequenties voor kinderen – te overwegen om DNA-onderzoek te doen in de oudst beschikbare generatie in de familie, omdat daar vaak minder/geen nieuwe verzekeringen aangegaan hoeven te worden en het eigen risicoprofiel, als er eenmaal gevorderd nierfalen is, niet erg meer zal veranderen met een genetische diagnose. De gestelde genetische diagnose helpt bij het adequaat informeren van familieleden en gerichte diagnostiek kan dan zo nodig worden ingezet.
Alle afdelingen klinische genetica (zijn academisch, maar via buitenpoliklinieken worden ook veel perifere ziekenhuizen bediend) leveren algemene nefrogenetische zorg (zie ook Poliklinieken klinische genetica).
Sinds 2015 zijn door de NFU expertisecentra voor zeldzame aandoeningen erkend, welke beschikbaar zijn voor overleg en verwijzingen. Zowel het UMC Utrecht (Expert Centre Hereditary and congenital nephrologic and urologic disorders), als het Radboudumc (Radboud Center Renal Disorders) heeft multidisciplinaire poliklinieken voor kinderen en volwassenen met complexe nierbeelden en/of een verdenking op een erfelijke nieraandoening. Bovendien zijn aan deze expertisecentra een multidisciplinair overleg (Radboud Center Renal Disorders) en een multidisciplinaire polikliniek zwangerschap en nieraandoeningen verbonden: voor pre-conceptieadvies, maar ook zwangerschapsbegeleiding (Polikliniek zwangerschap en nieraandoeningen UMC Utrecht). Het ErasmusMC heeft een multidisciplinair nefrogeneticaoverleg. Er zijn meer landelijk erkende expertisecentra op het gebied van specifieke zeldzame nieraandoeningen, de meest recente lijst staat op de website van Orphanet .
Begin 2017 is ERKNET, het Europese referentienetwerk voor zeldzame nieraandoeningen erkend door de Europese Unie (https://erknet.org). Binnen dit referentienetwerk kan informatie worden ingewonnen, bijvoorbeeld over individuele casus, van experts in die specifieke aandoening.
Overleg met of verwijzing naar een klinisch geneticus is altijd mogelijk. Het is belangrijk om de eventuele klinische implicaties mee te nemen in de afweging om wel of niet genetisch onderzoek te verrichten. Redenen voor contact kunnen zijn de verdenking op een erfelijke nieraandoening of urinewegafwijking, bijvoorbeeld op basis van:
In principe bij elke afwijkende bevinding, dus een VUS/klasse-3-variant of diagnose/klasse-4- of 5-variant*
In het algemeen is het aan te raden om counseling door een klinisch geneticus aan te bieden bij een (nieuw vastgestelde) erfelijke aandoening. Uiteraard kunt u ook patiënten verwijzen die opnieuw vragen hebben, bijvoorbeeld in het kader van kinderwens, of die de erfelijkheidsaspecten van hun diagnose niet volledig begrijpen.
Verder kunt u verwijzen bij:
Wanneer een nefroloog genetische diagnostiek heeft aangevraagd, is het in onderstaande gevallen in elk geval aan te raden naar een klinisch geneticus te verwijzen:
*Uitgaand van het adagium dat elke afwijkende uitslag een verwijsindicatie vormt, is deze opsplitsing enigszins arbitrair.
In bijlage 5 vindt u nuttige aanvullende literatuur en websites.
Degrees of genetic causality and power of molecular genetic diagnostic tests in monogenic and polygenic diseases
|
Recessive monogenic diseases |
Dominant monogenic diseases |
Polygenic diseases |
|
|
Genetic causality |
Strong |
Intermediate |
Weak |
|
Penetrance |
Full |
Sometimes incomplete |
Weak |
|
Predictive power of mutation analysis |
Almost 100% |
Strong* |
Weak |
|
Onset |
Fetus, child, adolescent |
Adult |
Adolescent, adult |
|
Molecular genetic approaches |
Direct exon sequencing of known disease genes |
Direct exon sequencing of known disease genes |
Only assignment of relative risk possible |
|
Frequency |
<1 in 40 000 (rare) |
<1 in 1000 (rare) |
<1 in 5 (frequent) |
|
Data usually derived from |
Gene mapping and gene identification |
Gene mapping and gene identification |
Genome-wide association studies |
|
Confirmation in animal model |
Very feasible |
Feasible |
Difficult |
*Except for incomplete penetrance and variable expression.
NB Overervingswijzen die hier niet genoemd worden zijn geslachtsgebonden en mitochondriële overerving.(2)
NB deze tabellen vormen een illustratie, maar zijn q.q. gedateerd en incompleet. Overgenomen uit Hildebrandt et al., 2010.(2) Zie ook Devuyst et al., 2014. (4)
Proteïnurie
|
Phenotype |
OMIM number |
MOI |
Characteristic signs and features |
Gene(s), gene product(s) |
|
SRNS: Congenital (Finnish type) |
256 300 |
AR |
Congenital nephrotic syndrome, CKD |
NPHS1, nephrin |
|
SRNS: Type 2 |
600 995 |
AR |
SRNS, FSGS, CKD |
NPHS2, podocin |
|
SRNS: Type 3 |
610 725 |
AR |
SRNS (SSNS), DMS, FSGS, CKD |
PLCE1, phospholipase C ε |
|
SRNS: Type 4 |
600 995 |
AR (AD) |
SRNS, FSGS |
CD2AP, CD2-associated protein |
|
SRNS: Pierson’s syndrome |
609 049 |
AR |
SRNS and microcoria |
LAMB2, laminin β2 |
|
SRNS: Adult-onset |
600 995 |
AD |
Adult-onset SRNS, FSGS, CKD |
ACTN4, α-actinin-4 |
|
SRNS: Adult-onset |
603 965 |
AD |
Adult-onset SRNS, FSGS, CKD |
TRPC6, transient receptor potential cation channel C6 |
|
Denys-Drash’s syndrome, Frasier’s syndrome |
194 080 |
AD |
Wilms’ tumour, pseudohermaphroditism, nephrotic syndrome |
WT1, WT suppressor gene 1 |
|
Nail-patella syndrome |
161 200 |
AD |
Nail dysplasia, absent patella, SRNS |
LMX1B, LIM homeodomain protein |
|
Schimke’s immuno-osseous dystrophy |
242 900 |
AR |
Bone abnormalities, immunodeficiency, SRNS |
SMARCAL1, HepA-related protein |
|
Mitochondrial disorders with SRNS |
607 426 |
AR |
SRNS with or without neurological impairment or SND |
COQ2, p-hydroxybenzoate- polyprenyltransferase; PDSS2, prenyl diphosphate synthase; MTTL1, mitochondrially encoded tRNA leucine 1 |
|
Lysosomal disorders with SRNS |
254 900 |
AR |
Action myoclonus, SRNS, CKD |
SCARB2, lysosomal integral membrane protein |
|
Glomerulopathy with fibronection deposits |
601 894 |
AD |
Proteinuria, distal RTA |
FN1, fibronectin 1 |
|
Alport’s syndrome |
301 050 |
XD |
Nephritis, SND, CKD |
COL4A5, α5(IV) collagen |
|
Alport’s syndrome with leiomyomatosis |
308 940 |
XD |
Alport’s syndrome with leiomyomatosis, CKD |
COL4A6, α6(IV) collagen |
|
Alport’s syndrome |
203 780 |
AR |
Alport’s syndrome or benign familial haematuria |
COL4A3, α3(IV) collagen |
|
Alport’s syndrome |
120 131 |
AR |
Nephritis, SND, CKD |
COL4A4, α4(IV) collagen |
|
OMIM=Online Mendelian Inheritance in Man database. MOI=mode of inheritance. SRNS=steroid-resistant nephrotic syndrome. AR=autosomal recessive. AD=autosomal dominant |
||||
|
CKD=chronic kidney disease. FSGS=focal segmental glomerulosclerosis. SSNS=steroid-sensitive nephrotic syndrome. DMS=diffuse mesangial sclerosis. SND=sensorineural deafness. RTA=renal tubular acidosis. XD=X-linked dominant. |
||||
Transabdominale echo: cysten, echodensiteit of tumor
|
Phenotype |
OMIM number |
MOI |
Characteristic signs and features |
Gene(s), gene product(s) |
|
ADPKD type 1 |
601 313 |
AD |
Polycystic kidneys, liver cysts, brain aneurysms, CKD |
PKD1, polycystin 1 |
|
ADPKD type 2 |
173 910 |
AD |
Polycystic kidneys, CKD |
PKD2, polycystin 2 |
|
ARPKD |
263 200 |
AR |
Polycystic kidneys, liver fibrosis, CKD |
PKHD1, fibrocystin and polyductin |
|
Nephronophthisis types 1-9 |
256 100 |
AR |
Polyuria, polydipsia, anaemia, CKD |
NPHP1 to NPHP9, nephrocystin types 1–9 |
|
Medullary cystic kidney disease |
174 000 |
AD |
Adult-onset CKD, hyperuricaemia, FJHN |
UMOD, Tamm–Horsfall protein |
|
Meckel-Gruber’s syndrome |
249 000, |
AR |
Polycystic kidneys, multiple-organ dysplasia, perinatal lethal |
MKS1; |
|
607 361 |
MKS3, meckelin (also allelic with NPHP genes) |
|||
|
Bardet-Biedl’s syndrome types 1–12 |
209 900 |
AR |
Retinitis pigmentosa, polydactyly, mental retardation, hypogenitalism, and obesity |
BBS1 to BBS12, Bardet-Biedl’s syndrome proteins |
|
Tuberous sclerosis type 1 |
191 100 |
AD |
Renal angiomyolipomas, skin changes, seizures |
TSC1, hamartin |
|
Tuberous sclerosis type 2 |
191 092 |
AD |
Renal angiomyolipomas, skin changes, seizures |
TSC2, tuberin |
|
von-Hippel-Lindau’s disease |
193 300 |
AD |
Lindau tumour, retinal angiomatosis, pheochromocytoma, renal tumour |
VHL, tumour suppressor gene g7 |
|
Wilms’-tumour-aniridia syndrome |
194 072 |
AD |
Wilms’ tumour, aniridia, growth retardation |
WT1, WT suppressor gene 1 |
|
Papillary renal cell carcinoma |
164 860 |
AD |
Papillary renal cell carcinoma |
MET, hepatocyte growth factor receptor |
|
OMIM=Online Mendelian Inheritance in Man database. MOI=mode of inheritance. ADPKD=autosomal dominant polycystic kidney disease. AD=autosomal dominant. |
||||
|
CKD=chronic kidney disease. ARPKD=autosomal recessive polycystic kidney disease. AR=autosomal recessive. FJHN=familial juvenile hyperuricaemic nephropathy. |
||||
Polyurie of tubulair verlies van elektrolyten, glucose, aminozuren, of overig metabool
|
Phenotype |
OMIM |
MOI |
Characteristic signs and features |
Gene symbol(s), gene product(s) |
Tubule segment expressing transporter or channel |
|
Renal glucosuria |
182 380 |
AR |
Renal glycosuria types A and B, glucose and galactose malabsorption |
SLC5A2, sodium-glucose cotransporter 2; |
Proximal |
|
233 100 |
SLC5A1, sodium-glucose cotransporter 1 |
||||
|
Proximal RTA |
259 730 |
AR |
Proximal RTA with extrarenal abnormalities |
CA2, carbonic anhydrase 2; |
Proximal tubule |
|
604 278 |
SLC4A4, sodium-bicarbonate cotransporter 4 |
||||
|
Hypophosphataemic rickets |
307 800, |
XD, |
Vitamin-D-resistant rickets with or without hyocalciuria |
PHEX, endopeptidase; |
Proximal tubule |
|
241 530 |
AR |
SLC34A3, sodium-phosphate cotransporter, (also FGF23, DMP1) |
|||
|
Bartter’s syndrome types 1–4 |
601 678 |
AR |
Hypokalaemic alkalosis, hypercalcuria, polyuria, growth retardation |
SLC12A1, sodium-potassium-chloridetransporter; |
mTAL |
|
241 200, |
CLCNKB, chloride channel Kb; |
||||
|
607 364 |
KCNJ1, potassium inwardly rectifying channel; |
||||
|
602 522 |
BSND; barttin |
||||
|
Gitelman’s syndrome |
263 800 |
AR |
Hypocalciuria, hypomagnesaemia, hypotension |
SLC12A3; thiazide-sensitive sodium-chloride cotransporter |
DCT |
|
Hypomagnesaemia |
248 250 |
AR |
Hypomagnesaemia, nephrocalcinosis, CKD, seizures |
CLDN16; claudin 16 |
DCT |
|
Hypomagnesaemia |
154 020 |
AD |
Hypomagnesaemia, seizures |
FXYD2, FXYD domain containing ion transport regulator 2 |
DCT |
|
Liddle’s syndrome |
177 200 |
AD |
PHA, hypertension |
SCNN1B, SCNN1G, sodium channel gain of function |
Collecting duct |
|
Gordon’s syndrome (PHA type 2) |
145 260 |
AD |
PHA type 2, increase in concentrations of potassium and chloride ions, acidosis, hypertension |
WNK4, WNK kinase 4; WNK1, WNK kinase 1 |
Collecting duct |
|
PHA type 1 |
264 350 (renal type) |
AD |
PHA type 1, concentration of sodium ions decreased, and potassium ions increased |
SCNN1A, SCNN1B, SCNN1G, sodium channel loss of function |
Collecting duct |
|
PHA type 1 |
264 350 (multiple type) |
AR |
PHA type 1, concentrations of sodium ions decreased, and potassium ions increased |
LCORL, mineralocorticoid receptor |
Collecting duct |
|
SeSAME syndrome |
612 780 |
AR |
Seizures, SND, ataxia, mental retardation, electrolyte wasting |
KCNJ1, potassium channel |
Collecting duct |
|
Distal RTA |
267 300 |
AR |
Distal RTA, nephrocalcinosis, SND, growth failure, osteomalacia |
ATP6B1, ATP6N1B,vacuolar ATPase units |
Collecting duct |
|
602 722 |
|||||
|
Distal RTA type 1 |
179 800 |
AD |
Distal RTA with haemolytic anaemia |
SLC4A1; erythrocyte band 3 (anion exchanger 1) |
Collecting duct |
|
Diabetes insipidus, nephrogenic |
304 800 |
XD |
Polyuria, polydipsia |
AVPR2, arginine vasopressin receptor 2; |
Collecting duct |
|
222 000 |
AR |
AQP2, aquaporin-2 |
|||
|
Cystinosis |
219 800 |
AR |
Renal Fanconi’s syndrome, photophobia, reduction in thyroxine concentrations |
CTNS, lysosomal membrane protein |
Secondary |
|
Lowe’s syndrome |
309 000 |
XR |
Cataract, vitamin D-resistant rickets, mental retardation, RTA, CKD |
OCRL1, phosphatidylinositol 4,5-bisphosphate 5-phosphatase |
Secondary |
|
Haemolytic uraemic syndrome, atypical |
235 400 |
AR |
Thrombocytopenia, haemolytic anaemia, acute renal failure |
CFH, complement factor H; CFHR1, complement factor H-related 1; CFHR3, complement factor, H-related 3; CD46; ADAMTS13 (AD) |
Secondary |
|
Fabry’s disease |
301 500 |
XR |
Angiokeratoma, FSGS, adult-onset CKD |
GLA, α-galactosidase A |
Secondary |
|
OMIM=Online Mendelian Inheritance in Man database. MOI=mode of inheritance. AR=autosomal recessive. RTA=renal tubular acidosis. XD=X-linked dominant. XR=X-linked recessive. |
|||||
|
mTAL=medullary thick ascending limb. DCT=distal convoluted tubule. CKD=chronic kidney disease. AD=autosomal dominant. PHA=pseudohypoaldosteronism. SeSAME=seizures, sensorineural deafness , ataxia, mental retardation, and electrolyte imbalance. SND=sensorineural deafness. Secondary=secondary tubulopathy due to cell damage. FSGS=focal segmental glomerulosclerosis. |
|||||
Nefrolithiasis / nefrocalcinose
|
Phenotype |
OMIM |
MOI |
Characteristic signs and features |
Gene symbol, gene product |
|
Cystinuria, type 1 |
220 100 |
AR |
Cystin calculi |
SLC3A1, solute carrier family 3 |
|
Cystinuria, non-type 1 |
604 144 |
AR |
Cystin calculi |
SLC7A9, solute carrier family 7 |
|
Dent’s disease |
300 009 |
XR |
Nephrolithiasis, nephrocalcinosis, renal Fanconi’s syndrome |
CLCN5, renal chloride channel |
|
Primary hyperoxaluria type 1 |
259 900 |
AR |
Nephrolithiasis, CKD |
AGXT, alanineglyoxylate aminotransferase |
|
Primary hyperoxaluria type 2 |
260 000 |
AR |
Nephrolithiasis |
GRHPR, glyoxylate reductase |
|
Lysinuric protein intolerance |
222 700 |
AD |
Nephrolithiasis, phosphate wasting, osteopenia |
SLC9A3R1, solute carrier family 9 |
|
Adenine-phosphoribosyl- transferase deficiency |
102 600 |
AR |
Nephrolithiasis |
APRT, adenine phosphoribosyl transferase |
|
Xanthinuria |
278 300 |
AR |
Nephrolithiasis, xanthine calculi |
XDH, xanthine dehydrogenase |
|
Distal renal tubular acidosis |
179 800 |
AD |
Nephrolithiasis, rickets |
SLC4A1, solute carrier family 4 |
|
OMIM=Online Mendelian Inheritance in Man database. MOI=mode of inheritance. AR=autosomal recessive. XR=X-linked recessive. |
||||
|
CKD=chronic kidney disease. AD=autosomal dominant. |
||||
CAKUT (congenital anomalies of the kidney and urinary tract)
|
Phenotype |
OMIM |
MOI |
Renal features |
Extrarenal features |
Gene(s), product(s) |
|
CAKUT |
601 090 |
AD |
CAKUT |
Iridiodysgenesis |
FOXC1, forkhead transcription factor C1 |
|
Renal agenesis |
191 830 |
AD |
Renal agenesis, adysplasia, vesicoureteral reflux |
Allelic with MEN2A, facial defects |
RET, ret proto-oncogene; |
|
611 559 |
UPK3A, uroplakin 3A |
||||
|
RHD |
112 262 |
AD |
RHD |
Microphthalmia, cleft lip |
BMP4, bone morphogenetic protein 4; |
|
604 994 |
SIX2, sine oculis 2 |
||||
|
Multicystic renal dysplasia |
602 868 |
AD |
Multicystic renal dysplasia |
– |
CDC5L, cell division cycle 5; |
|
600 390 |
USF2, upstream transcription factor 2 |
||||
|
Vesicoureteral reflux |
602 431 |
AD |
Vesicoureteral reflux |
Subtle facial and limb defects |
ROBO2, roundabout 2; |
|
603 746 |
SLIT2, slit homologue 2 |
||||
|
Branchio-otorenal syndrome |
601 653 |
AD |
CAKUT, RHD, vesicoureteral reflux |
Deafness, ear malformation, branchial cysts |
EYA1, eyes absent homologue 1; |
|
159 980 |
MYOG, myogenin; |
||||
|
601 205 |
SIX1, sine oculis 1; |
||||
|
600 963 |
SIX5, sine oculis 5 |
||||
|
Fraser’s syndrome |
607 830 |
AR |
Renal agenesis, RHD |
Cryptophthalmos-syndactyly syndrome |
FRAS1, extracellular matrix protein; |
|
608 945 |
FREM2, FRAS1-related extracellular matrix protein |
||||
|
Hypoparathyroidism, deafness, renal syndrome |
146 255 |
AD |
CAKUT |
Hypoparathyroidism, deafness, and renal defects |
GATA3, GATA binding protein 3 |
|
Kallman’s syndrome |
308 700 |
AD |
Renal agenesis |
Anosmia, hypogenitalism |
KAL1, anosmin |
|
Renal coloboma syndrome |
167 409 |
AD |
CAKUT (vesicoureteral reflux, RHD) |
Retinal coloboma |
PAX2, paired box gene 2 |
|
Renal cysts and diabetes syndrome, GCKD |
137 92 |
AD |
RHD, cysts |
Maturity-onset diabetes of the young type 5 diabetes, genital anomalies, GCKD |
HNF1B, transcription factor 2 |
|
609 886 |
|||||
|
SHFM |
603 273 |
AD |
Urethral malformation |
SHFM |
BMP7, bone morphogenetic protein 7; |
|
600 028 |
DLX5, distal-less homeobox 5; |
||||
|
600 030 |
DLX6, distal-less homeobox 6; |
||||
|
603 273 |
TP63, tumour protein p63 |
||||
|
Townes-Brocks’ syndrome |
107 480 |
AD |
Renal agenesis, RHD |
Limb, ear, anal abnormalities |
SALL1, sal-like 1 |
|
OMIM=Online Mendelian Inheritance in Man database. MOI=mode of inheritance. AD=autosomal dominant. RHD=renal hypodysplasia. AR=autosomal recessive. |
|||||
|
GCKD=glomerulocystic kidney disease. SHFM=split-hand and split-foot malformation. |
|||||
|
Phenotype |
Synonyms |
Genes |
link to FACD |
|
Birt-Hogg-Dubé syndrome |
BHD, Fibrofolliculomas with Trichodiscomas and Acrochordons, incl.: Hornstein-Knickenberg syndrome, Perifollicular Fibromatosis |
FLCN |
|
|
von Hippel-Lindau disease |
VHL |
VHL |
|
|
Hereditary Papillary Renal Carcinoma (type 1) |
HPRC, Hereditary Papillary Renal Cell Cancer |
MET, MITF |
|
|
Hereditary Leiomyomatosis and Renal Cell Cancer |
HLRCC, Reed syndrome, Hereditary Multiple Leiomyomata of Skin and Uterus |
FH |
|
|
Familial Wilms’ tumor |
|
FWT1/WT4#, FWT2#, WT1 |
|
|
Wilms’ tumor-Aniridia-ambiguous Genitals-mental Retardation |
WAGR syndrome |
cgd(11p), PAX6, WT1 |
|
|
Tuberous Sclerosis |
TS, Tuberous Sclerosis Complex, TSC, Bourneville-Pringle disease |
TSC1, TSC2 |
|
|
Cowden syndrome |
CS, Cowden disease, Multiple Hamartoma Syndrome, incl.: Lhermitte-Duclos; part of PTEN hamartoma tumour syndrome (PHTS) / PTEN-MATCHS, Cowden-like syndrome |
ATK1, KILLIN, PIK3CA, PTEN, SDHB, SDHD |
|
|
Familial Paragangliomas |
Hereditary Glomus Tumors, Familial Paragangliomas, Hereditary Paragangliomas, type 1-3: PGL1, PGL2, PGL3, incl. Familial Carotid Body Paraganglioma and Sensorineural Hearing Loss |
SDHA, SDHAF2/SDH5, SDHB, SDHC, SDHD |
Preïmplantatie genetische diagnostiek (PGD; “embryoselectie”) wordt in Nederland sinds 1995 aangeboden. Het is één van de alternatieven die paren met een verhoogde kans op een kind met een monogene aandoening hebben voor het invullen van hun kinderwens.
Het betreft een IVF/ICSI procedure waarbij een embryo zonder de familiaire aanleg wordt geselecteerd middels genetisch onderzoek op één cel van een blastomeer (drie dagen na bevruchting).
PGD is alleen mogelijk als er eerst een zekere genetische diagnose gesteld is. Bij nefrogenetische aandoeningen is dit laatste steeds beter mogelijk.
Voordat PGD wordt gestart is eerst goedkeuring voor de aandoening in kwestie nodig van de nationale indicatiecommissie. Aanvankelijk werd PGD alleen verricht bij paren met een verhoogd risico op een kind met een ernstige lichamelijke aandoening en of verstandelijke beperking op de baby- of kinderleeftijd. In recentere jaren zijn echter ook aandoeningen met een volwassen debuut en variabele expressie goedgekeurd. Dit, in combinatie met de toegenomen aantal geïdentificeerde genen voor nieraandoeningen en de toegenomen PGD capaciteit, betekent dat PGD kan worden aangeboden aan paren met een verhoogde kans op een kind met een nieraandoening, ook met debuut op volwassen leeftijd.
Tot en met 2015 zijn 2762 paren gestart met een PGD procedure, waarvan in totaal 58 vanwege een erfelijke nieraandoening. In totaal zijn er 85 cycli uitgevoerd, die geleid hebben tot 21 doorgaande zwangerschappen. Er zijn meer paren verwezen (n=143) dan gestart met een cyclus. (www.PGDNederland.nl)
De meeste paren die zijn verwezen waren drager van een mutatie in PKD1. Enkele andere voorbeelden zijn de ziekte van Alport (n=17) en nefrogene diabetes insipidus (n=6), maar ook hereditaire FSGS door een INF2 mutatie (n=2) of de ziekte van Wilson (n=1). Tevens zijn medullaire cystenieren door een MUC1 mutatie een reden geweest om met patiënten een verwijzing naar het PGD-team aan te bieden.
De verwijzing voor PGD gaat altijd via een klinisch geneticus. Het genetisch onderzoek op één cel wordt verricht in het Maastricht UMC+, de nationale vergunninghouder. Het UMCG, UMC Utrecht en het AMC zijn transportcentra, waar patiënten de IVF procedure kunnen doorlopen. Deze vier centra werken samen in “PGD Nederland”. Ook klinisch genetici in andere centra kunnen met patiënten een eerste informerend gesprek over PGD hebben en hen desgewenst daarna verwijzen.
Paren met een erfelijke nieraandoening dienen tijdig geïnformeerd te worden over de verschillende alternatieven omtrent het invullen van kinderwens, waaronder PGD, omdat het (van het beginnen met aantonen van een genetische diagnose tot aan een eerste terugplaatsing) lang kan duren. Patiënten met interesse in/vragen over het invullen van hun kinderwens kunnen verwezen worden naar een klinisch geneticus. Een minderheid van de paren kiest uiteindelijk voor PGD.
Boek
Genetic Diseases of the Kidney, eerste editie 2009, Edited by: Richard P. Lifton, Stefan Somlo, Gerhard H. Giebisch and Donald W. Seldin, ISBN: 978-0-12-449851-8
Websites
Patiënteninformatie (ook nuttig voor verwijzers)
Klinische (nefro-)genetische zorg en diagnostiek
Medisch inhoudelijk
* Niet expliciet genoemd in dit document
© 2022. Alle rechten voorbehouden